Calling loops from a sample
This workflow demonstrates how to call loops using AQuA-tools in a Docker or Tinker environment.
We will use the following AQuA tools:
Motivation
In the previous recipe, we focused our analysis on HiChIP contacts between known linear 1D peaks derived from prior ChIP-seq data. However, very often we do not have paired ChIP experiments, and it becomes necessary to call meaningful interactions directly from the contact matrix itself. In such cases, obtaining a visually appealing set of loop calls is critical before performing any downstream operation for hypothesis testing.
In this recipe, we’ll use two independent cell lines:
sample1: H3K27ac HiChIP performed on the cell line GM12878
sample2: H3K27ac HiChIP performed on the cell line K562
For both experiments, we do not have paired linear 1D peaks from H3K27ac ChIP-seq experiments.
Chapter 0: Ingredients
I. Prerequisites
You must complete the AQuA-tools Docker installation before starting, which can be found here. Alternatively, if you have access to Tinker, AQuA-tools are already pre-installed and no installation is needed.
II. Creating a workspace
This is an optional step. We encourage users to organize their analyses into a directory structure that keeps things nice and clean.
working_directory=/home/ubuntu/container_outputsmkdir -p $working_directorycd $working_directorymkdir -p \ data \ scripts \ results \ scratchIt’s nice to have all the files you want to play with in $working_directory/data
For now, we’ll follow the steps of this tutorial by housing ourselves in $working_directory/scratch, but we recommend writing .sh scripts in $working_directory/scripts and storing the results in $working_directory/results
III. Gathering necessary files
For downstream analysis, we will need a TAD file and a TSS file. We’ll assign these file paths to variable names for easier calling later.
cd $working_directory/scratch
# Initialise files in variablesTAD=/home/ubuntu/lab-data/hg38/reference/TAD_goldsorted_span_centromeres-removed_hg38.bed
TSS=/home/ubuntu/lab-data/hg38/reference/GENCODE_TSSs_hg38.bedIV. Find a genomic region of interest
Now that we have our TAD and TSS files, we can choose a locus to use in our downstream analysis. For this recipe, we will use bedtools intersect to find the TAD that contains the MYC locus, but you can use this process for any gene of interest to you.
# Get coordinates for MYC locus from TSS file# NOTE: all gene names are in upper casegrep -w MYC $TSS > MYC.bedcat MYC.bed# chr8 127735434 127735435 MYC +
# Get coordinates of the TAD that contains MYCbedtools intersect -a $TAD -b MYC.bed -wa -wb# chr8 126817755 129115254 8 chr8 127735434 127735435 MYC +The first three columns are the range of the TAD where MYC resides, which we will use as the genomic range to tinker with when calling loops. When specifying a range to AQuA tools, the required format is chr:start:end
# Storing TAD coordinates in a variablemyc_tad=$(bedtools intersect -wa -a "$TAD" -b <(awk '$4=="MYC" {print}' "$TSS") | awk '{print $1 ":" $2 ":" $3}')
echo $myc_tad# chr8:126817755:129115254V. Defining a sample
We’ll need a sample to get started. In Tinker or Docker environments, you’ll use the name of the sample as it appears on the Tinker box or Docker container.
# on Tinker or Dockersample=GM12878_H3K27acNOTE: You can list all loaded samples on your tinkerbox or Docker container using list_samples
Chapter 1: Calling loops for a region of interest
The loop-calling tool in the AQuA-tools suite is extract_bedpe. extract_bedpe scans all possible bin‐pairs within a defined region (or regions, think TADs), and outputs those .bedpe pairs with contact scores above a user‐defined inherent score and distance threshold. To know more about inherent scores, check out our Docs.
I. Extracting the .bedpe
In this example, we’ll use extract_bedpe to scan the sample and region defined in Chapter 0. This recipe covers several key features of the tool, but we also recommend checking out our Docs to learn more about how it works.
extract_bedpe \ --sample1 $sample \ --range $myc_tad \ --genome hg38 > GM12878_MYC.bedpeII. Viewing the .bedpe
We can visualize the output of extract_bedpe with the AQuA-tool plot_contacts. This tool accepts a .bedpe file via the --bedpe parameter, which will highlight the regions in the .bedpe on the contact plot. For more information about how plot_contacts works, see our Docs.
plot_contacts \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --bedpe GM12878_MYC.bedpe \ --output_name MYC.pdfOn Tinker, you can view this pdf by downloading it from the file browser. On Docker, you can open the file directly from your local output directory.
Here’s our pdf! The plot on the left uses default parameters, including the CPM normalization color scale. The plot on the right is the same plot but with the inherent normalization color scale. To know more about inherent normalization, check out our Docs.
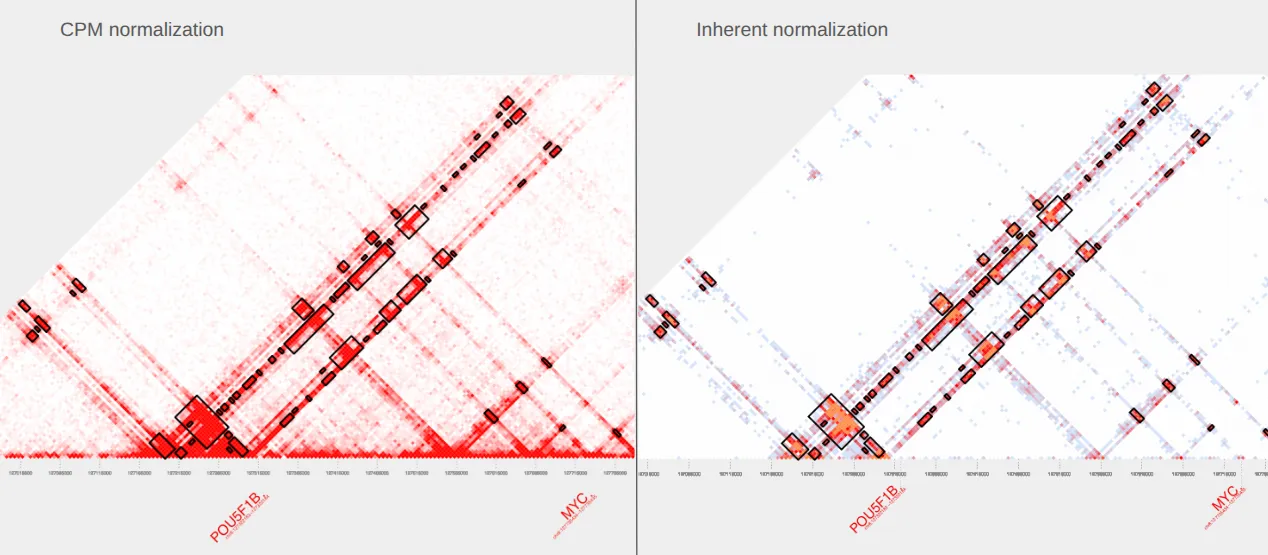
Chapter 2: Changing extraction parameters
I. Changing the extract_bedpe threshold score
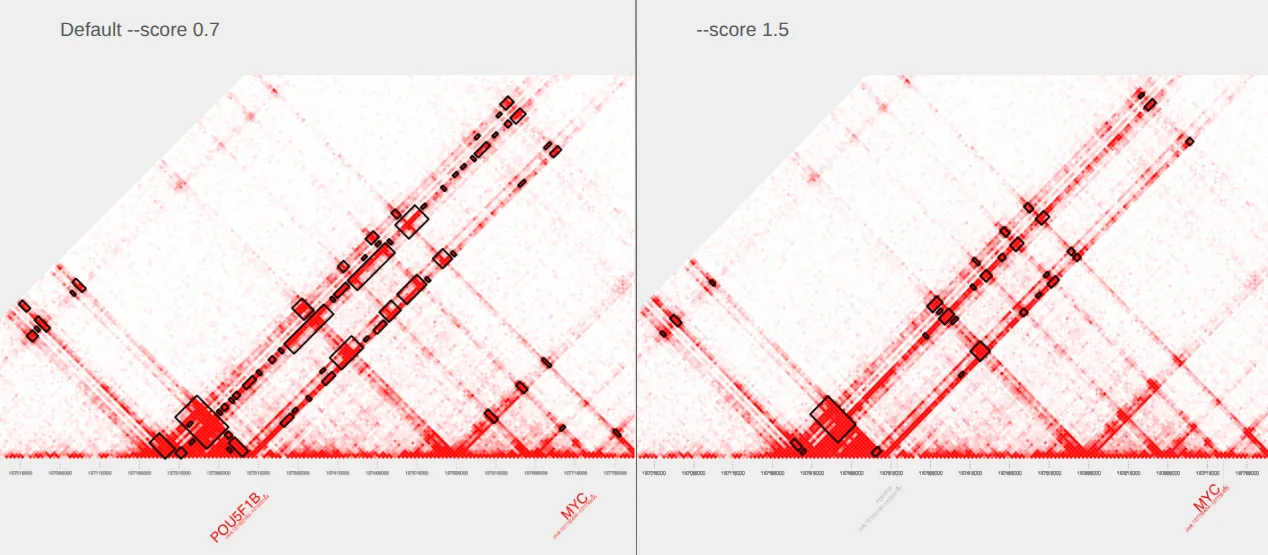
In this step, we’ll re-run the same extract_bedpe command from Chapter 1, but with an additional --score parameter to set a custom score threshold. This allows us to control which loops are extracted based on contact strength. Since extract_bedpe uses inherent normalization, most loop scores fall between 0 and 1. The default threshold is 0.7, but by increasing it to 1.5, we focus on loops with the strongest contact signals.
extract_bedpe \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --score 1.5 > GM12878_MYC_score-1.5.bedpeWe can view the output of extract_bedpe using plot_contacts once again:
plot_contacts \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --bedpe GM12878_MYC_score-1.5.bedpe \ --output_name MYC_score-1.5.pdfThe plot on the left is our original plot with default parameters and the plot on the right shows our updates. In the plot on the right, notice how the highlighted .bedpe regions now align more closely with the strongest loop structures, and appear slightly smaller and more focused than in the default plot on the left.
II. Changing the extract_bedpe mode
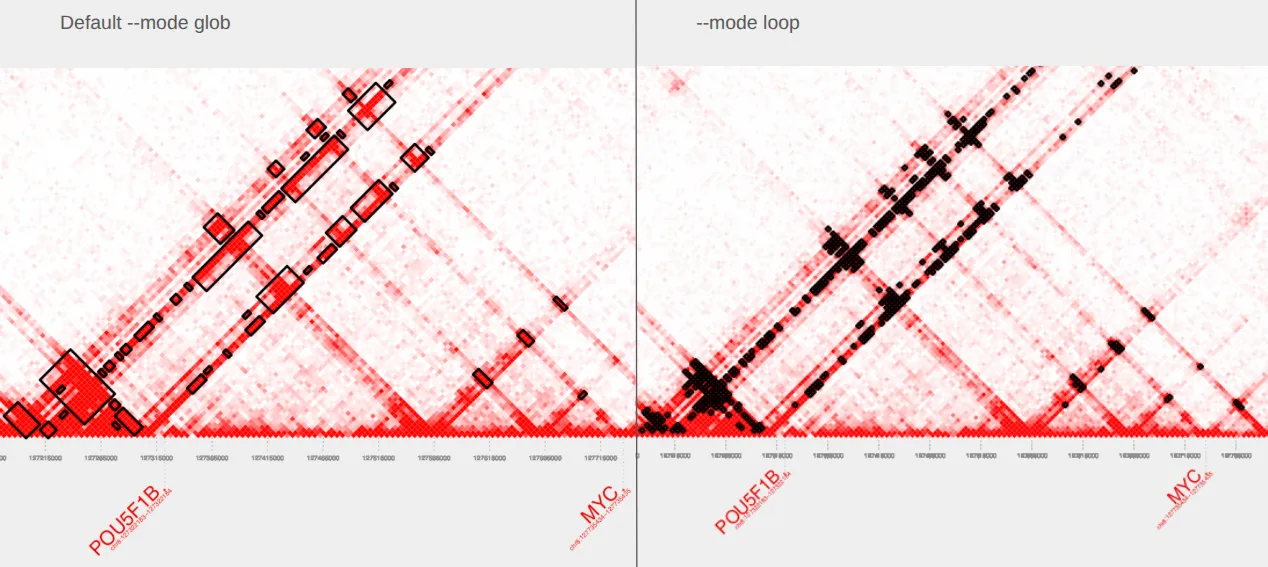
—mode glob
The default mode for extract_bedpe is --mode glob, which requires loops to span at least two consecutive bins. This helps eliminate loops that may be noise, and is the mode we have been using by default so far.
—mode loop
To view individual loops, we’ll use --mode loop with extract_bedpe and then plot with plot_contacts.
extract_bedpe \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --mode loop > GM12878_MYC_loop.bedpe
plot_contacts \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --bedpe GM12878_MYC_loop.bedpe \ --output_name MYC_loop.pdfThe plot on the left is our original plot with default parameters and the plot on the right shows our extracted bedpe with --mode loop specified.
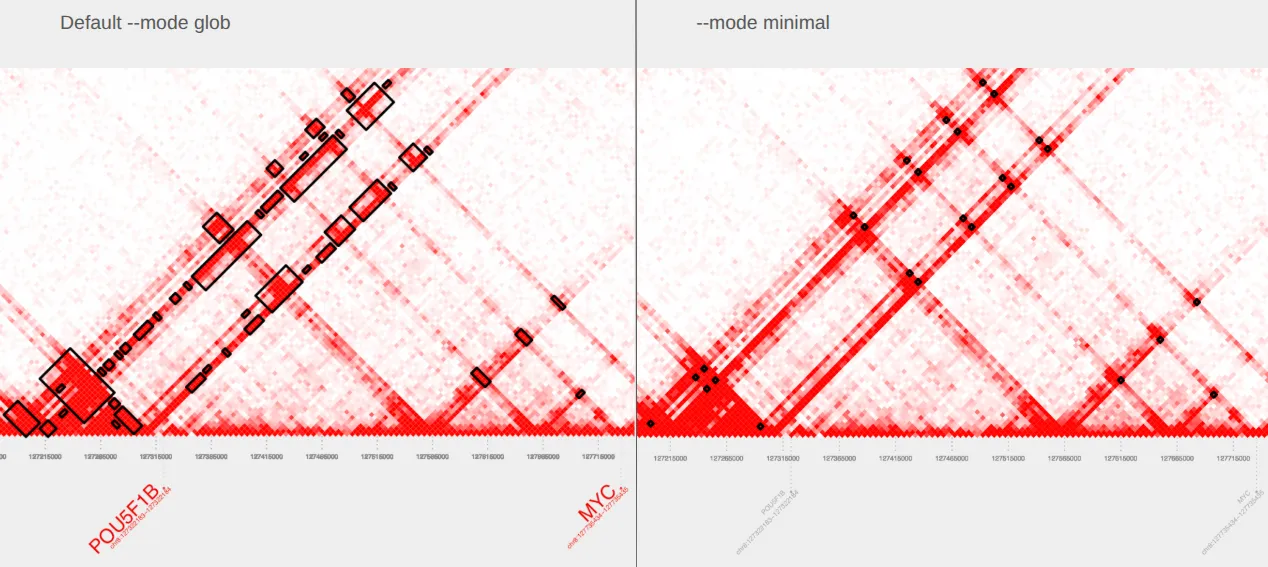
—mode minimal
With --mode minimal, extract_bedpe detects the strongest interaction anchors based on both signal strength and interconnectivity. This produces a minimal loop set containing the most important features in the interaction network.
extract_bedpe \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --mode minimal > GM12878_MYC_minimal.bedpe
plot_contacts \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --bedpe GM12878_MYC_minimal.bedpe \ --output_name MYC_minimal.pdfCompare our original plot with default parameters on the left to the plot with --mode minimal specified on the right.
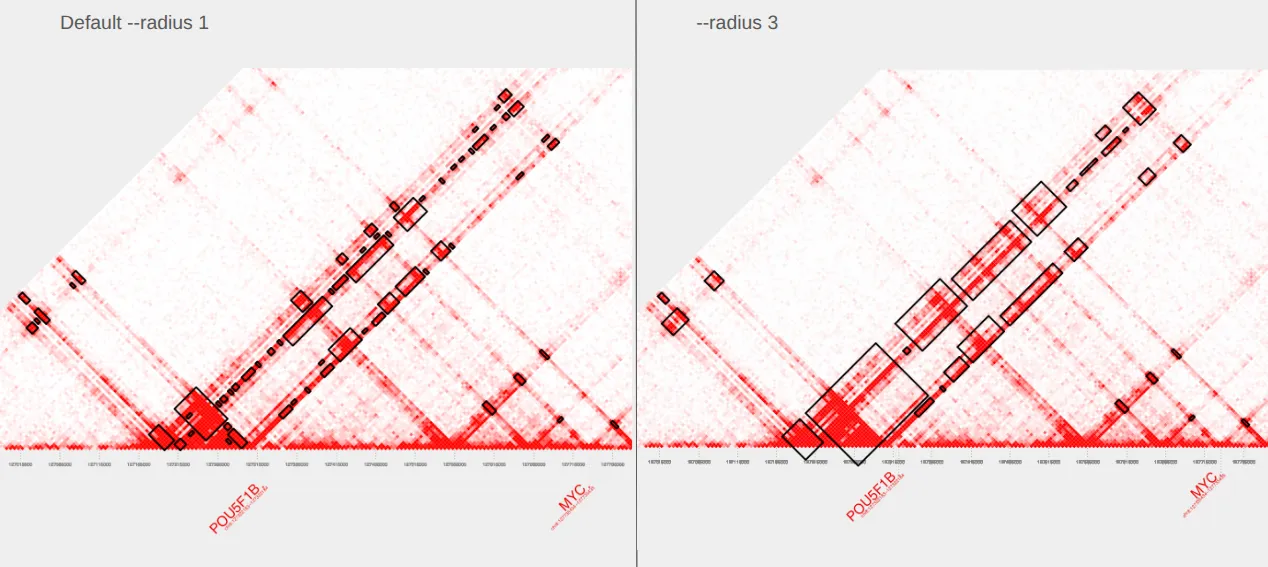
III. Changing the extract_bedpe radius
When using --mode glob or --mode flare, the --radius parameter determines the gap between reported regions of interaction. The default --radius is 1, but can be increased to join more distant bins. Here we compare .bedpe files made with the default --radius 1 and an increased --radius 3.
extract_bedpe \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --radius 3 > GM12878_MYC_radius-3.bedpe
plot_contacts \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --bedpe GM12878_MYC_radius-3.bedpe \ --output_name MYC_radius-3.pdfCompare our original plot with default parameters on the left to our updated plot with --radius 3 specified on the right. Notice how this conglomerates nearby loops.
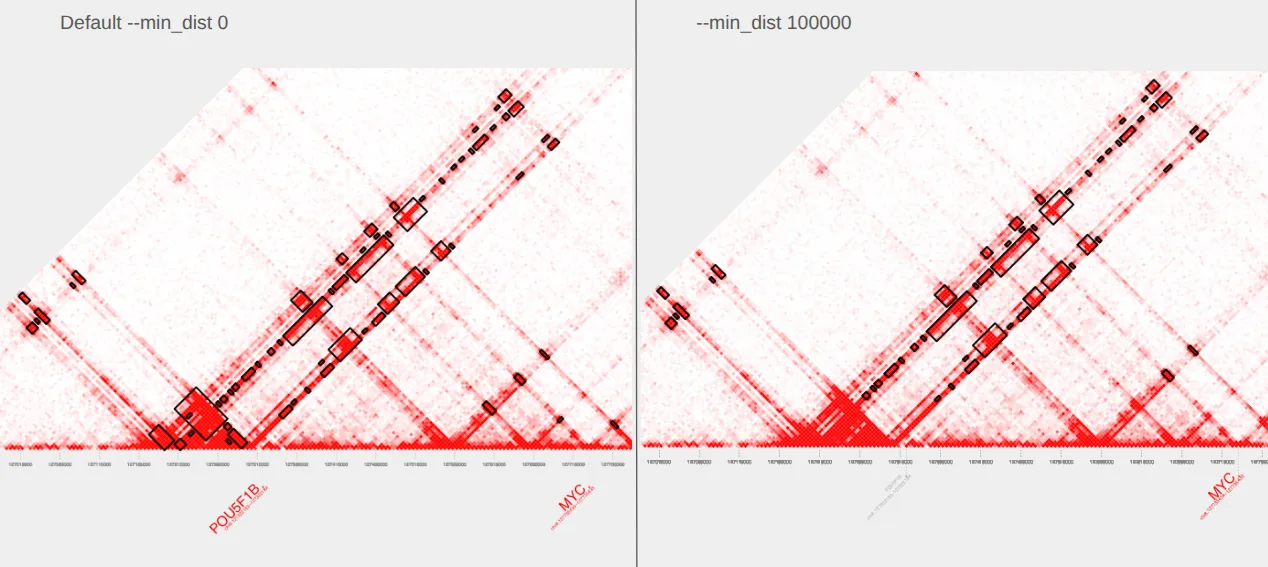
IV. Changing the extract_bedpe min_dist
To exclude loops close to the diagonal, we can supply a --min_dist value to extract_bedpe. Here, we filter pairs that are separated by less than 100,000 bp and plot.
extract_bedpe \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --min_dist 100000 > GM12878_MYC_mindist-100kb.bedpe
plot_contacts \ --sample1 $sample \ --range $myc_tad \ --genome hg38 \ --bedpe GM12878_MYC_mindist-100kb.bedpe \ --output_name MYC_mindist-100kb.pdfNotice the loss of loops nearest the diagonal in the plot on the right compared to the original plot with default parameters on the left.
Chapter 3: Genome-wide Loop Calling
I. Extracting the genome-wide .bedpe
extract_bedpe can be used to call loops across the entire genome, although this process is slower than the examples in previous chapters using a defined range for a locus, like MYC. Calling loops genome-wide can take up to 30 minutes. If you’re working on Docker, you have access to pre-computed genome-wide loop files in the directory /home/ubuntu/genome-wide_loops and you can skip this step.
# Genome wide loop calling
extract_bedpe \ --sample1 GM12878_H3K27ac \ --genome hg38 \ --TAD $TAD \ --min_dist 20000 > GM12878_genome-wide-loops.bedpe
extract_bedpe \ --sample1 K562_H3K27ac \ --genome hg38 \ --TAD $TAD \ --min_dist 20000 > K562_genome-wide-loops.bedpeWe’ll be using the output from this step in the next recipe.
Conclusion
In this recipe, you learned how to call chromatin loops using extract_bedpe and visualize them with plot_contacts in a Docker or Tinker environment. Starting from a single genomic region, we walked through how to fine-tune loop calling parameters like score thresholds, modes, radius, and minimum distance.
This approach provides a way to explore HiChIP contact maps directly, without predefined annotations like enhancers, promoters, or TADs. Our goal is to make tailored loop calling easy for your biological question, whether you’re zooming in on a gene like MYC or scanning the entire genome for large-scale interaction patterns. What might extract_bedpe uncover in your data?
In the next recipe, we’ll build on the results from this recipe by integrating genome-wide loop calls into comparative analyses. But before moving on, feel free to experiment with other regions, scores, and visualization styles to get a feel for how AQuA-tools works with your data.