extract_bedpe
Concepts
The aqua_tools suite provides two primary methods for gathering data for analysis:
-
build
If you are starting with known regions in a BED file, such as promoters or enhancers,
build_bedpecan convert these regions into paired-end regions for 3D contact analysis. -
extract
If you are uncertain about the specific regions you need,
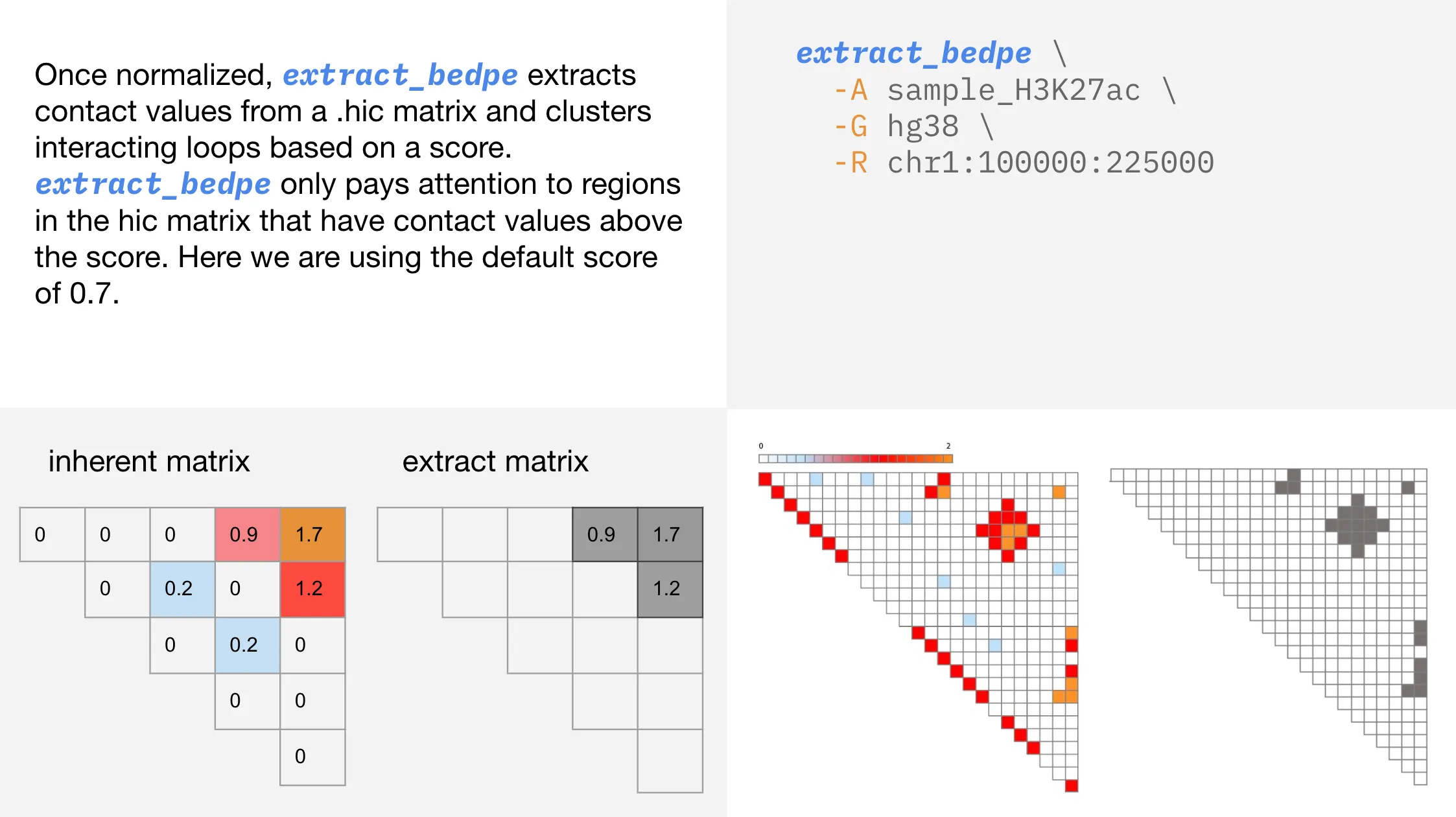
extract_bedpeallows you to analyze contact regions without prior knowledge of their genomic elements or biological significance, making it ideal for exploratory research. By starting with a .hic file,extract_bedpeidentifies regions in contact, regardless of whether they are enhancers, promoters, or other genomic elements.
In this tutorial we will cover the basics of extract_bedpe and explain its customizable parameters. extract_bedpe is a flexible tool which will allow you to tailor your analysis to focus on the most relevant contacts, filtering out noise and improving signal clarity.
Using inherent normalization

Click here to learn more about inherent normalization.
Extracting contacts from a .hic
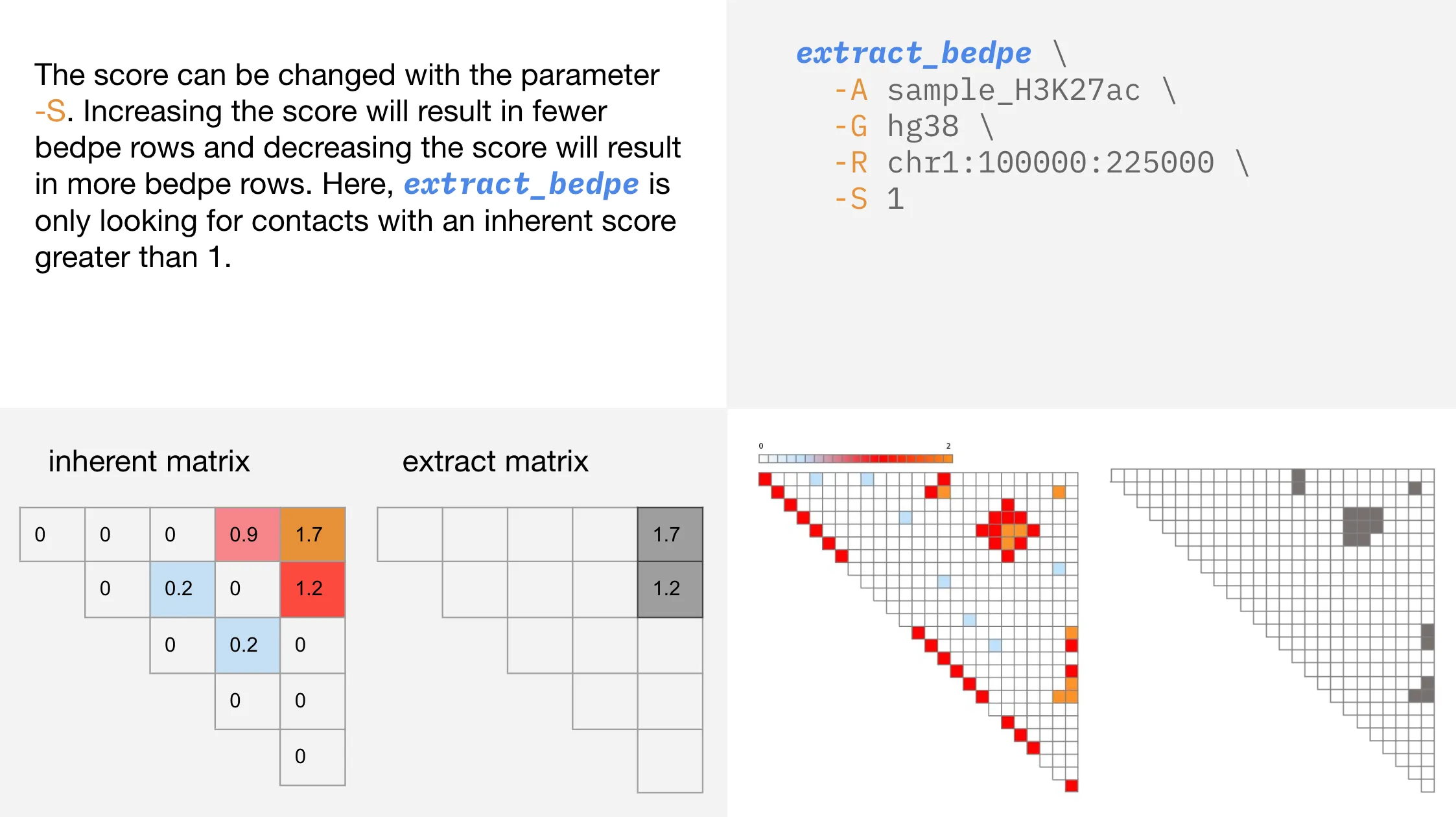
Changing the contact score
You select the level of contact that extract_bedpe considers, allowing for quick identification of the strongest contacts within a region.

Minimum distance constraint
Once you have obtained regions with the desired level of contact, you may want to further narrow your search. Using the --min_dist parameter, you can filter your results to ignore regions that are within a certain distance from the diagonal (indicating they are close together in linear space).

Understanding your output
To illustrate the meaning of the output from extract_bedpe, we have drawn shadows for 4 example regions to show which regions the output refers to.

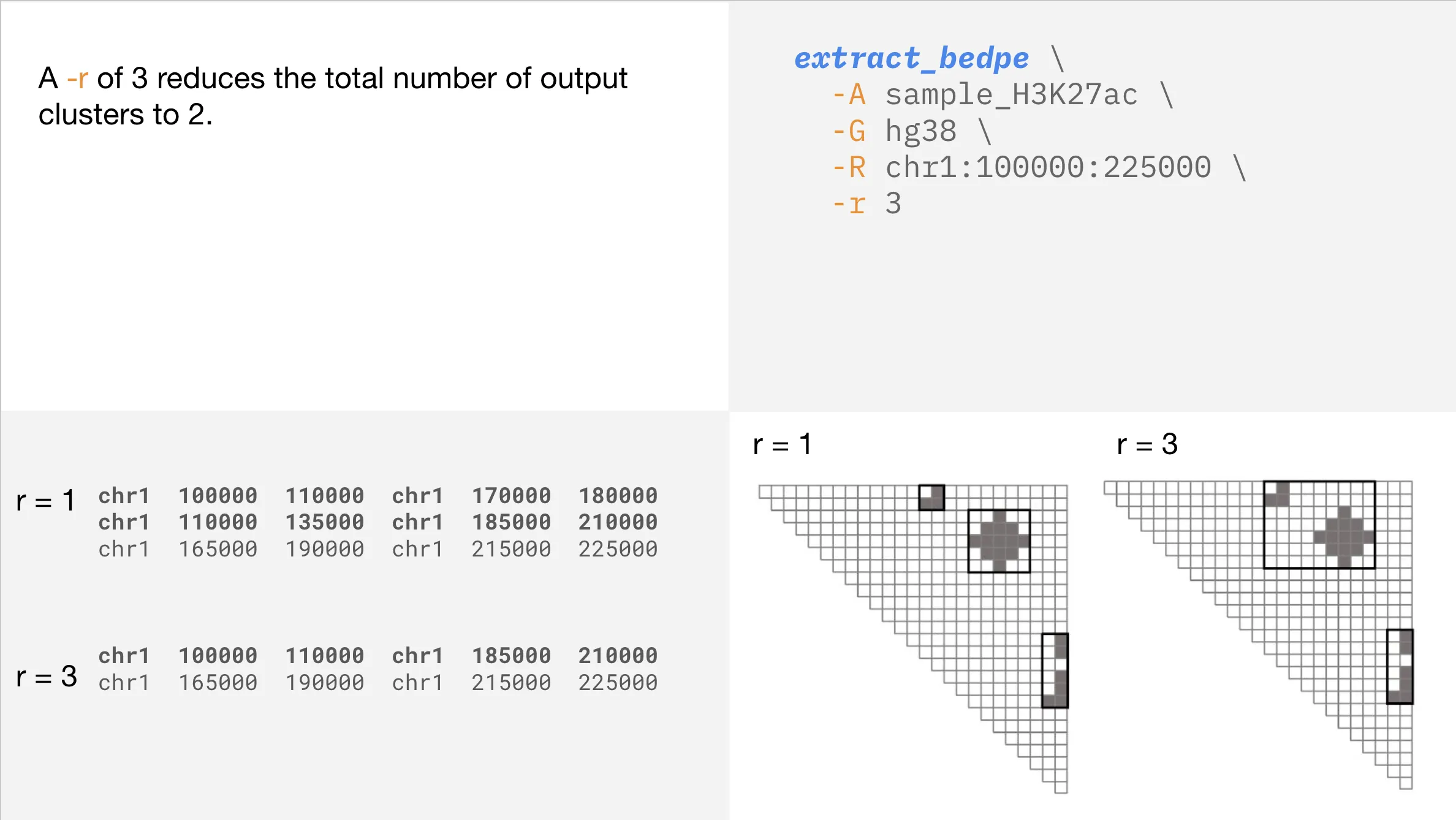
Conglomerating regions
Refining your output is easy with additional parameters like --radius. If your output seems too granular, you can use this parameter to conglomerate nearby loops.

Changing the extract_bedpe —mode



Searching within a range
Depending on your analysis, you may only be interested in contacts (whether they’re loops or globs) within a specific range.

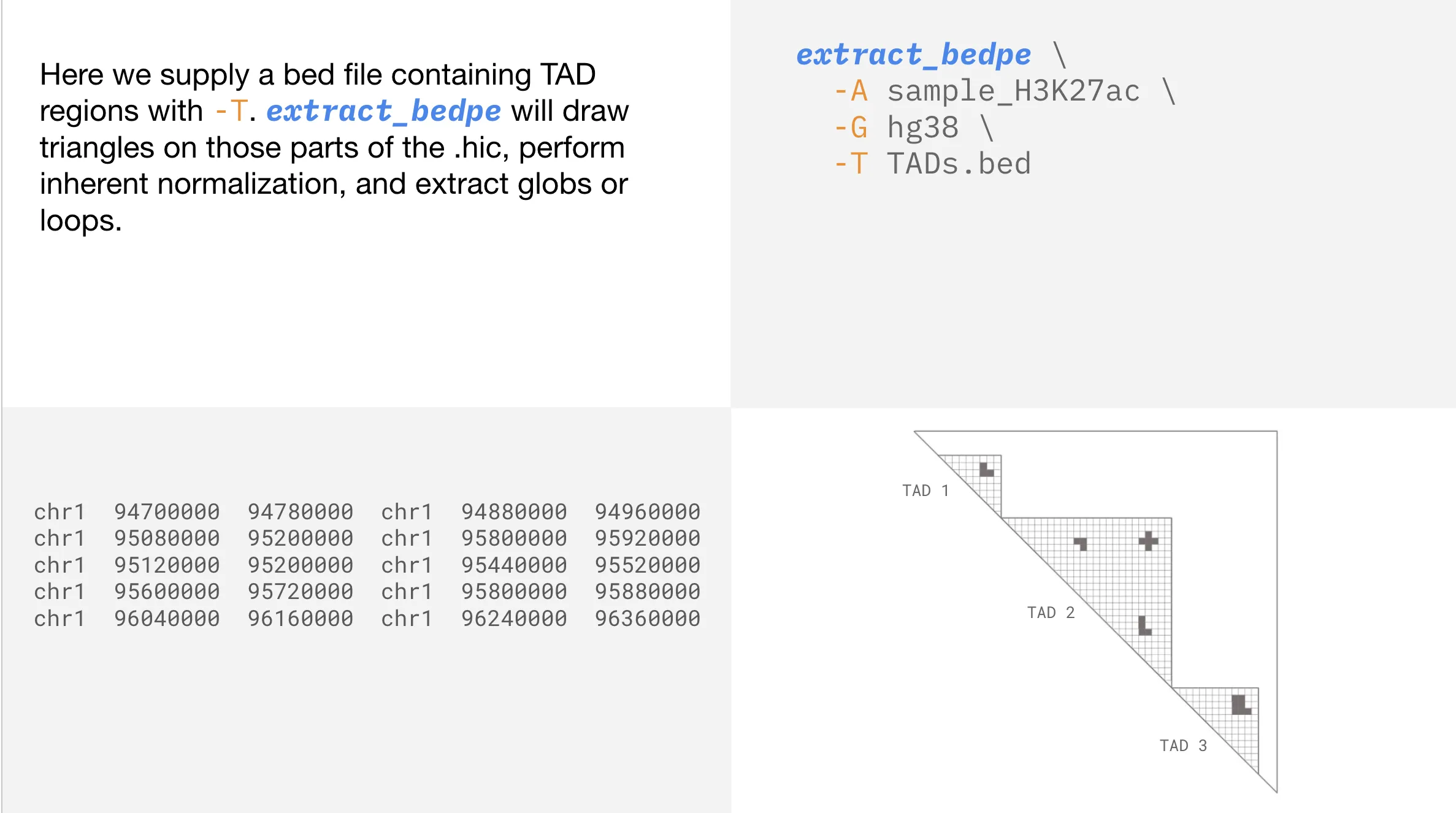
Searching within TAD boundaries
If you have a bed file of TADs, or any other type of boundaries, you can tell extract_bedpe to only consider contacts within those boundaries.
Usage
extract_bedpe obtains clusters of interacting loops based on a score threshold. Scores are calculated using inherent normalization and printed in bedpe format to standard out.
Usage and Option Summary
extract_bedpe -A sample -G genome -R rangeRequired
| Short Option | Long Option | Description |
|---|---|---|
-A | --sample1 | Name of sample on Tinkerbox |
-G | --genome | Genome build the sample was processed using |
-R | --range | Range to obtain clusters from, in chr:start:end format |
Optional
| Short Option | Long Option | Description |
|---|---|---|
-r | --resolution | Resolution in base pairs. Only 5000 and 1000 supported. Default 5000 |
-T | --TAD | Full path to TAD file, the boundaries of which will be used to obtain clusters |
-S | --score | Inherent score to seed cluster formation. Default = 1 |
-m | --mode | Shape of bedpe to be called. Strictly loop, flare, minimal, or glob. Default glob |
--radius | Bin distance units to search for neighbours . Default 1 | |
--min_dist | Distance in basepairs to filter out extracted elements . Default = 0 | |
-h | --help | Help message |