query_bedpe
Concepts
For many AQuA tool users, query_bedpe serves as the go-to secondary tool in the pipeline when addressing questions related to genomic interactions. After you’ve obtained a .bedpe file with build_bedpe, query_bedpe is the next logical step to focus on the interactions that matter most for your research.
Visualize bedpe
Let’s explore some key features and settings that make query_bedpe a versatile tool for 3G genomics research. We’ll start by visualizing an interaction in a .bedpe file.

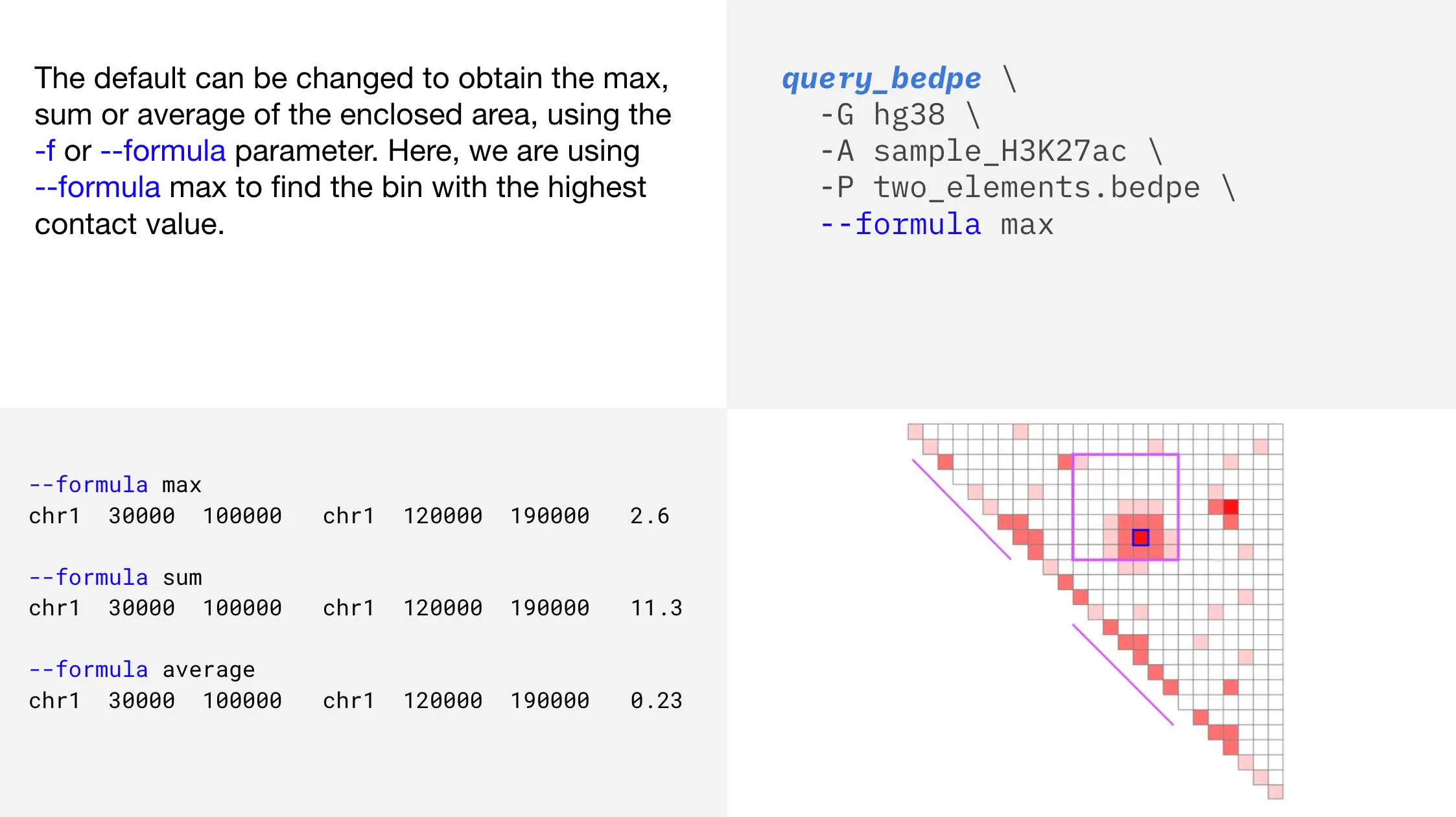
Report contact values
The purple square shows the bedpe region, and the two diagonal purple lines show the bedpe feet. Now, let’s use query_bedpe to determine contact values within our bedpe. The contact value will be printed to standard out in the 7th column.

In the above plot, we can see that the strongest interaction (represented by the darkest color of red), is not the value that is reported when we use the default query_bedpe settings.
That’s better! Now, how do we get those coordinates?
Update coordinates

Usage
query_bedpe uses a bedpe file to calculate AQuA normalized or counts-per-million (CPM) contact values for given ranges in a sample and prints to standard out.
Usage and Option Summary
query_bedpe -A H3K27ac -P path/to/pairs.bedpe -G hg38(or):
query_bedpe -A H3K27ac -B H3K27me3 -P path/to/pairs.bedpe -G hg38Required
| Short Option | Long Option | Description |
|---|---|---|
-P | --bedpe | Full path to the bedpe (pairs) file you want to query, without headers! |
-A | --sample_1 | Name of the sample you want to use as it appears on the Tinker box |
-G | --genome | The genome build the sample(s) has been processed using. Strictly hg19 or hg38 |
Optional
| Short Option | Long Option | Description |
|---|---|---|
-Q | --norm | Which normalization to use. Strictly ‘none’, ‘cpm’, ‘aqua’, or ‘abc’ in lower case. Non-spike-in samples default to cpm. Spike-in samples default to aqua |
-B | --sample_2 | The name of the second sample. If triggered, calculates the delta contact values for that pair. Useful in case vs control |
-r | --resolution | Resolution of sample in base pairs. Default 5000. Accepted resolutions: 1000,5000,10000,25000,50000,100000,250000,500000,1000000,2500000 |
-f | --formula | Arithmetic to use to report contact values. Options: center, max, average, sum. Default = center |
-F | --fix | If FALSE, reports new coordinates based on arithmetic center or max. Default = TRUE |
--expand | Expands 1D bedpe feet in both directions based on supplied value (in bin units). Default = 0 | |
-i | --inherent | If TRUE, hic values transformed to inherent units. For one-sample tests only. Default = FALSE |
-m | --preserve_meta | If TRUE, bedpe metadata columns will be preserved. Default = TRUE |
-c | --cores | Number of cores to use, default is determined automatically |
-h | --help | Help message |