Visualizing Loops
This code snippet shows how to visualize loops with plot_contacts in a Docker or Tinker environment.
We’ll use the same sample we used to call loops in the previous snippet.
sample=GM12878_H3K27acWe’ll also define the genome build and range of interest.
genome=hg38range=chr2:64500000:65000000Here we assign the path to our loops file to a variable:
loops=my_loops.bedpeAnd let’s give a name for our output .pdf:
name=contact_plot.pdfFirst, let’s run plot_contacts to look at our contact plot without our loops highlighted.
plot_contacts --sample1 $sample --genome $genome --range $range --output_name $nameHere’s our pdf:
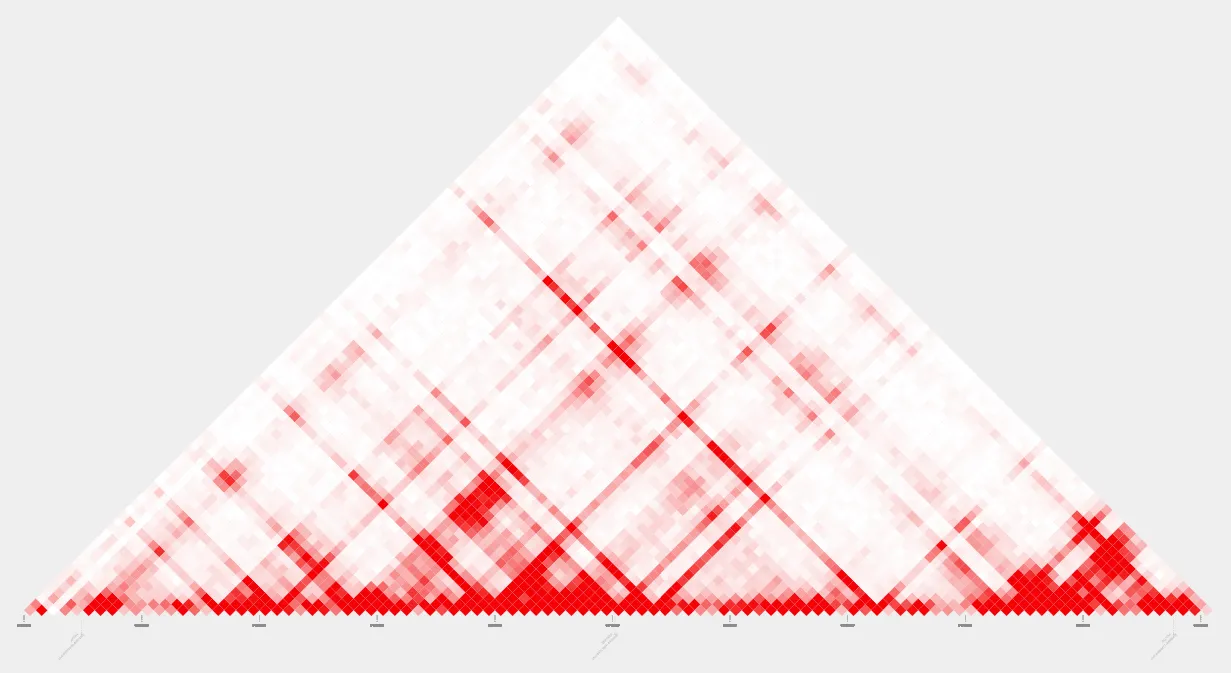
Next, let’s run the same code again with the addition of the --bedpe parameter to highlight our loops. We’ll also update the output name for this plot:
name=my_loops.pdfplot_contacts --sample1 $sample --genome $genome --range $range --bedpe $loops --output_name $nameHere’s our pdf:
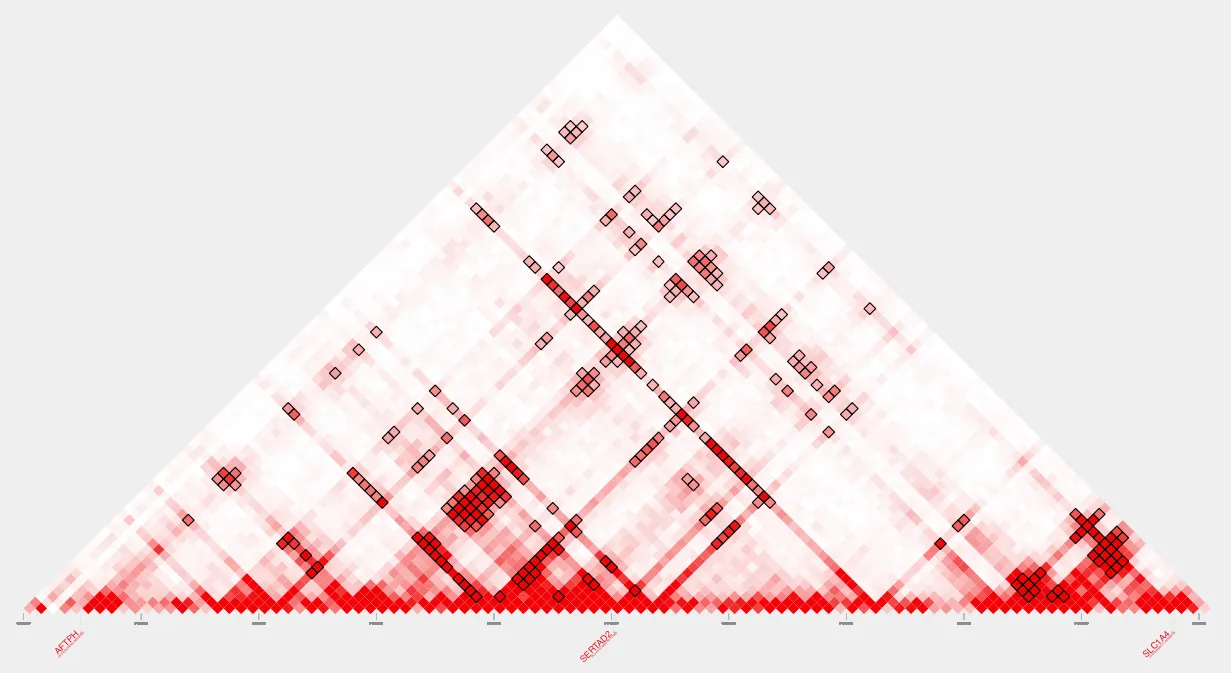
To apply Inherent Normalization, set the parameter --inherent to TRUE.
name=my_loops_inh.pdfplot_contacts --sample1 $sample --genome $genome --range $range --bedpe $loops --output_name $name --inherent TRUEHere’s our inherent normalized plot:
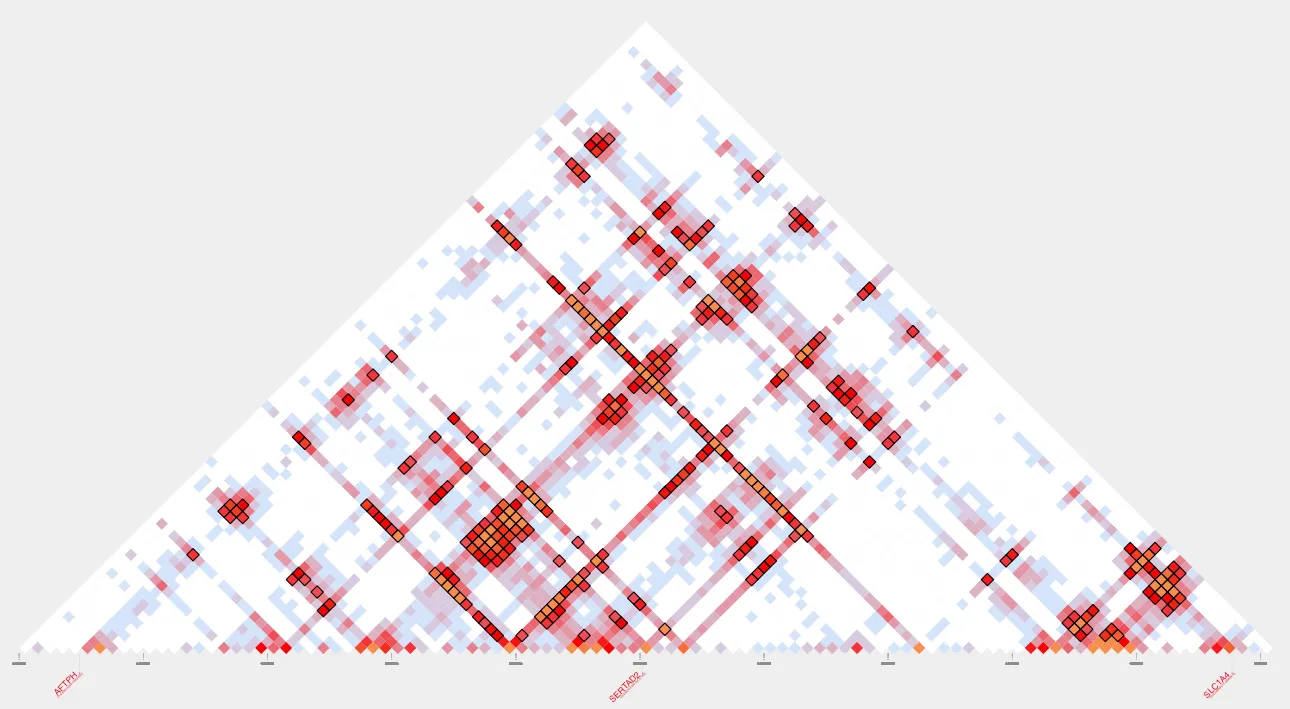
To intersect these loops with other regions of interest, please continue to the next Snippet: Intersecting Loops